Sản xuất MSC-EV cho lâm sàng: Vai trò của môi trường CD, AOF và GMP
Môi trường CD, AOF đang dần trở thành chủ đề được quan tâm hàng đầu trong sản xuất MSC và MSC-EV, đặc biệt khi các yêu cầu về GMP và thẩm định lâm sàng ngày càng khắt khe. Không ít khách hàng đặt câu hỏi: môi trường chemical defined (CD), animal-origin-free (AOF) là gì, khác gì so với môi trường nuôi MSC truyền thống có huyết thanh, và liệu có bắt buộc phải sử dụng môi trường CD/AOF trong quy trình sản xuất MSC-EV hay không? Việc hiểu đúng bản chất và vai trò của môi trường CD, AOF chính là bước đầu tiên để xây dựng quy trình nuôi cấy ổn định, an toàn và sẵn sàng cho ứng dụng lâm sàng.
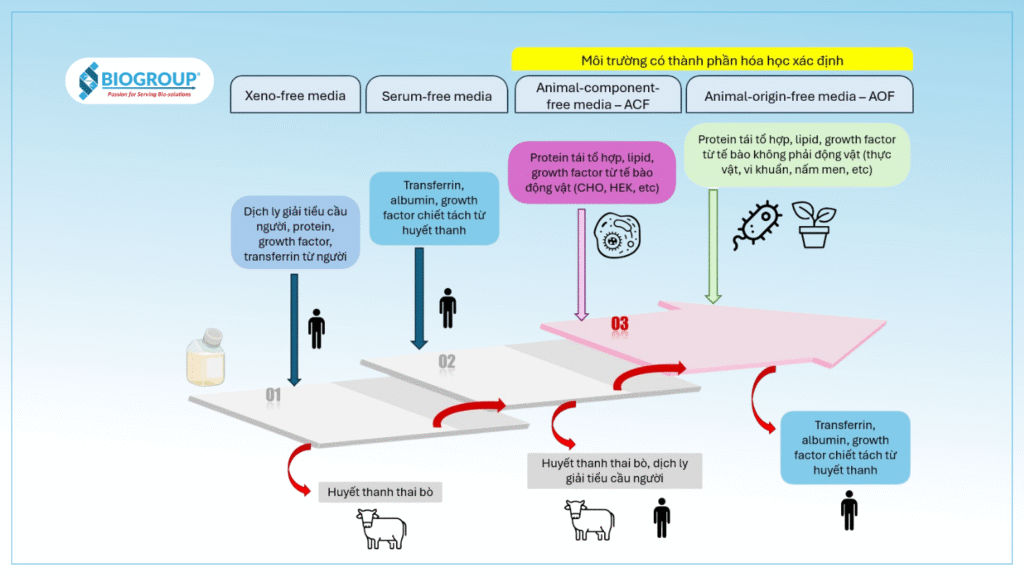
Định nghĩa môi trường CD, AOF
Về mặt định nghĩa, môi trường có thành phần hóa học xác định (chemically defined – CD) là môi trường nuôi cấy mà tất cả các thành phần hóa học đều được định danh và định lượng chính xác. Trong khi đó, tiêu chuẩn không có nguồn gốc động vật (Animal-Origin-Free – AOF) đảm bảo quy trình sản xuất hoàn toàn không chứa các dẫn xuất từ động vật và người. Sự kết hợp này giảm thiểu nguy cơ nhiễm tạp ngoại lai và các tác nhân gây bệnh có nguồn gốc động vật đến mức thấp nhất, đồng thời tăng tính ổn định giữa các lô sản xuất.
Dù về mặt hiệu suất tăng trưởng thuần túy, môi trường CD/AOF có thể không mang lại sự bứt phá vượt bậc so với môi trường có bổ sung huyết thanh, nhưng chúng lại là điều kiện cần thiết về mặt pháp lý, đáp ứng được các tiêu chuẩn GMP và vượt qua các rào cản kiểm duyệt lâm sàng.
Nhằm giải quyết vấn đề trên, môi trường CellCor™ MSC CD AOF đạt chuẩn GMP đã ra đời, phục vụ tăng sinh tế bào. Bên cạnh đó, dòng CellCor™ EXO CD, là môi trường chuyên dụng để thu hồi túi ngoại bào (EV). Với ưu điểm độ tinh khiết cao và không tạp nhiễm các hạt nhỏ, CellCor™ EXO CD là lựa chọn tối ưu cho quá trình nuôi cấy và tách chiết EV hạ nguồn.
Giải thích một số nhãn thường thấy trong các môi trường nuôi cấy (1,2)
Môi trường không chứa huyết thanh (Serum-free media)
Môi trường hoàn toàn loại bỏ thành phần huyết thanh hoặc huyết tương. So với môi trường truyền thống, các dòng serum-free media mang lại sự ổn định vượt trội giữa các lô sản xuất (batch-to-batch consistency), đồng thời cho phép người dùng kiểm soát chặt chẽ hơn các điều kiện nuôi cấy
Môi trường không có nguồn gốc ngoại lai (Xeno-free media)
Môi trường không chứa các thành phần có nguồn gốc từ động vật không phải người. Tuy nhiên, môi trường này có khả năng chứa các thành phần từ người như huyết thanh người (human serum), holo-transferrin, hoặc insulin.
Môi trường không chứa protein (Protein-free media)
Môi trường không chứa protein hoặc polypeptides trong thành phần công thức. Môi trường này có thể chứa amino acids tự do, dipeptides hoặc tripeptides chiết xuất từ nguồn không phải động vật. Có thể chứa dịch thủy phân từ thực vật, nấm men hoặc vi khuẩn.
Môi trường có thành phần hóa học xác định (chemical defined media – CD)
Loại môi trường nuôi cấy tế bào chứa các thành phần hữu cơ và vô cơ tinh khiết, cùng các protein, vitamin, axit béo và các yếu tố tăng trưởng đã được định lượng rõ ràng.
Môi trường không chứa thành phần động vật (Animal-component-free media – ACF)
Môi trường không chứa hoặc không sử dụng bất kỳ nguyên liệu thô sơ cấp nào có nguồn gốc trực tiếp từ mô hoặc dịch cơ thể động vật (bao gồm cả người) trong quá trình sản xuất. Môi trường có thể chứa các protein động vật tái tổ hợp, bao gồm cả những loại được sản xuất từ các dòng tế bào động vật hoặc thông qua quá trình lên men.
Ví dụ: môi trường có chứa protein người tái tổ hợp được sản xuất từ tế bào thận phôi người (HEK).
Môi trường không có nguồn gốc từ động vật (Animal-origin-free media – AOF)
Sản phẩm cuối cùng không chứa và không sử dụng bất kỳ nguyên liệu thô sơ cấp hay thứ cấp nào có nguồn gốc trực tiếp từ mô hoặc dịch cơ thể động vật (bao gồm cả người) trong quá trình sản xuất. Các nguyên liệu thô tam cấp có thể được dẫn xuất từ mô, dịch cơ thể hoặc các dòng tế bào động vật (bao gồm cả người).
Ví dụ: môi trường chứa protein người tái tổ hợp được sản xuất và chiết xuất từ gạo.
Môi trường đạt chuẩn GMP
Là môi trường được sản xuất dựa trên quy trình mà tất cả sản phẩm, nguyên liệu, thiết bị và công đoạn vận hành đều có khả năng truy xuất nguồn gốc và được kiểm soát chặt chẽ.
Sự cần thiết của môi trường nuôi cấy có thành phần hóa học xác định cho nuôi cấy quy mô công nghiệp
Trong sản xuất công nghiệp, tính ổn định của từng mẻ nuôi cấy là tiêu chí cốt lõi để đảm bảo chất lượng và sự đồng nhất của sản phẩm đầu ra. Việc sử dụng các thành phần không xác định trong môi trường nuôi cấy luôn tiềm ẩn rủi ro làm sụt giảm sản lượng và chất lượng bất thường (3). Điều này không chỉ gây lãng phí nguồn lực mà còn gây khó khăn lớn cho quá trình điều tra và khắc phục sự cố cũng như tối ưu hóa quy trình.
Một minh chứng khác nằm ở việc sử dụng huyết thanh thai bò (FBS). Ngay cả khi đến từ cùng một nhà sản xuất, các lô FBS khác nhau vẫn có sự biến thiên lớn về thành phần, gây ảnh hưởng trực tiếp đến khả năng tăng sinh của tế bào. Nghiên cứu của Zhang và cộng sự trên dòng tế bào biểu mô sắc tố võng mạc người trưởng thành (ARPE) đã chỉ ra rằng: ba lô FBS khác nhau từ hai nhà cung cấp cho tốc độ phát triển tế bào hoàn toàn khác biệt (4). Mặc dù hàm lượng protein chính tương đương nhau, nhưng lô FBS cho tốc độ tăng sinh nhanh nhất lại chứa các yếu tố tăng trưởng và protein liên kết đặc hiệu mà các lô còn lại không có (4). Sự khác biệt khó kiểm soát này chính là rào cản lớn nhất trong việc chuẩn hóa các quy trình nuôi cấy hiện nay.
Hiện nay, nhiều nghiên cứu MSC vẫn đang sử dụng môi trường truyền thống là DMEM bổ sung 10% serum (từ bò hoặc từ người). Hệ thống này có thể phù hợp ở quy mô phòng thí nghiệm hoặc khi mới thiết lập quy trình ban đầu, nhưng sẽ bộc lộ nhiều hạn chế nếu muốn tăng quy mô sản xuất và kiểm soát chặt chẽ đầu vào
Thực tế, đã có nhóm nghiên cứu chuyển sang dùng hPL (dịch ly giải tiểu cầu người) để thay thế FBS. Tuy nhiên, khi bắt đầu nâng quy mô, họ đã gặp phải một điểm nghẽn lớn là tế bào không tăng sinh nhanh như dự đoán. Sau khi điều tra, nhóm mới phát hiện lô hPL mới đã bị nhiễm mycoplasma. Sự cố này không chỉ gây gián đoạn mà còn khiến họ mất tới vài tháng để khắc phục và làm sạch hệ thống.
Do đó, việc chuyển dịch sang môi trường Chemically Defined (CD) chính là giải pháp để tránh những rủi ro bất ngờ như trên. Môi trường CD đảm bảo sự ổn định tuyệt đối giữa các mẻ nuôi cấy, giúp nhà nghiên cứu hoàn toàn kiểm soát được quy trình, từ đó đơn giản hóa việc theo dõi và tối ưu hóa sản xuất ở quy mô lớn.
Môi trường không có nguồn gốc từ động vật (AOF): Ưu thế và xu hướng
Ngành công nghiệp sinh học đã trải qua nhiều bài học đắt giá: từ thảm họa nhiễm mycoplasma (thập niên 70), nỗi lo về endotoxin (thập niên 80) cho đến cuộc khủng hoảng bò điên BSE (thập niên 90). Chính những cột mốc này đã hình thành nên tiêu chuẩn khắt khe về việc truy xuất nguồn gốc nguyên liệu ngày nay.
Để ngăn ngừa những rủi ro tương tự trên diện rộng, FDA và ICH đã đưa ra các hướng dẫn nghiêm ngặt nhằm kiểm soát tính an toàn của sinh phẩm và hoạt chất. Cụ thể, theo hướng dẫn ICH Q5A(R2) về đánh giá an toàn virus cho các sản phẩm sinh học, các nhà sản xuất được khuyến cáo (5):
Ưu tiên thay thế: Các nhà sản xuất nên tránh sử dụng nguyên liệu nguồn gốc từ người và động vật (như huyết thanh, trypsin…) trong quy trình sản xuất bất cứ khi nào có thể.
Minh bạch hồ sơ: Nếu bắt buộc phải sử dụng, nhà sản xuất phải cung cấp đầy đủ dữ liệu về quốc gia xuất xứ, loại mô nguồn gốc, các bước bất hoạt virus hoặc quy trình loại bỏ virus được áp dụng trong quá trình sản xuất nguyên liệu, cũng như các loại xét nghiệm virus đã được thực hiện trên nguyên liệu thô đó đều phải được cung cấp đầy đủ. Các xét nghiệm này nên được thực hiện trước giai đoạn bất hoạt.
Kiểm soát bổ sung: Trong trường hợp có nguy cơ cao, các nguyên liệu như huyết thanh hoặc trypsin phải được xử lý bất hoạt virus (như chiếu xạ ion hóa). Ngoài ra, nhà sản xuất cần áp dụng thêm các chiến lược giảm thiểu rủi ro như lọc virus, xử lý nhiệt cao trong thời gian ngắn hoặc chiếu xạ UVC để đảm bảo an toàn tuyệt đối.
Việc sử dụng môi trường chứa thành phần từ động vật hoặc người thường dẫn đến gánh nặng về thủ tục hành chính khi lập hồ sơ đăng ký sản phẩm. Do đó, môi trường không có nguồn gốc động vật (AOF) đang trở thành lựa chọn ưu tiên của các đơn vị sản xuất nhờ những lợi thế vượt trội:
Kiểm soát an toàn: Loại bỏ các rủi ro sinh học khó lường và nguy cơ nhiễm tạp virus.
Tối giản hồ sơ: Giảm áp lực về việc chứng minh nguồn gốc và kiểm soát virus trước các cơ quan quản lý (FDA/ICH).
Tiêu chuẩn hóa: Đáp ứng hoàn hảo các yêu cầu khắt khe trong quản lý chất lượng dược phẩm hiện nay.
Dù nhiều quy trình ban đầu vẫn sử dụng môi trường chứa thành phần từ động vật ở giai đoạn nghiên cứu, việc thiết lập lộ trình chuyển đổi sang AOF nên được thực hiện ngay từ sớm. Điều này giúp doanh nghiệp tránh được việc phải tái thiết kế quy trình tốn kém và mất thời gian khi bước vào giai đoạn sản xuất lâm sàng.
GMP là tiêu chuẩn gì? Khi nào thì cần chuyển đổi sang GMP?
Định nghĩa về cGMP
CGMP là các quy định về Thực hành tốt sản xuất hiện hành (current good manufacturing practice) do FDA giám sát và thực thi. CGMP cung cấp một hệ thống nhằm đảm bảo việc thiết kế, giám sát và kiểm soát quy trình sản xuất cũng như cơ sở hạ tầng được thực hiện một cách phù hợp. Việc tuân thủ các quy định cGMP giúp bảo đảm danh tính, hoạt lực, chất lượng và độ tinh khiết của các sản phẩm thuốc, thông qua yêu cầu các nhà sản xuất dược phẩm phải kiểm soát chặt chẽ mọi hoạt động vận hành sản xuất (10).
Tại sao cGMP lại là tiêu chuẩn quan trọng
Thực tế, người tiêu dùng không thể nhận biết một sản phẩm thuốc có an toàn hay hiệu quả hay không chỉ bằng cảm quan thông thường. Dù việc kiểm nghiệm sản phẩm là bắt buộc, nhưng chỉ riêng khâu này là không đủ để đảm bảo chất lượng. Thông thường, việc kiểm soát chỉ được thực hiện trên một mẫu nhỏ (ví dụ: test 100 viên trong lô 2 triệu viên), nhằm giữ lại phần lớn sản phẩm phục vụ bệnh nhân thay vì tiêu hủy hết cho việc thử nghiệm. Do đó, điều cốt yếu là thuốc phải được sản xuất trong các điều kiện và quy trình nghiêm ngặt theo quy định CGMP, nhằm đảm bảo chất lượng được tích hợp vào từng bước của quy trình thiết kế và sản xuất (10).
Khi nào thì cần chuyển đổi sang GMP
Theo quy định từ FDA và EMA, các sản phẩm là thuốc hoặc liệu pháp tế bào đều bắt buộc phải đáp ứng tiêu chuẩn GMP trước khi được cấp phép sử dụng trên người (6–8). Ngay cả đối với giai đoạn thử nghiệm lâm sàng, FDA hiện cũng đã ban hành hướng dẫn cụ thể về tiêu chuẩn cGMP cho các sản phẩm sử dụng ngay từ giai đoạn 1, bao gồm cả giả dược (9).
Dựa trên các quy định này, trong giai đoạn nghiên cứu cơ bản tại phòng thí nghiệm, quy trình có thể chưa cần tuân thủ hoàn toàn các tiêu chuẩn GMP khắt khe. Tuy nhiên, ngay khi sản phẩm được định hướng sử dụng trong các thử nghiệm lâm sàng hoặc điều trị trên người, việc nâng cấp và chuyển đổi sang quy trình sản xuất đạt chuẩn cGMP là yêu cầu bắt buộc để đảm bảo tính pháp lý và an toàn cho đối tượng sử dụng.
Do đó, trong quá trình nghiên cứu, nên dùng môi trường có phiên bản GMP ngay từ giai đoạn R&D nếu có ý định đi xa, để tránh việc phải làm lại từ đầu các thí nghiệm kiểm chứng khi đổi môi trường ở giai đoạn sau.
Môi trường GMP có ý nghĩa gì
Môi trường nuôi cấy đóng vai trò then chốt cho các sản phẩm liệu pháp tế bào, như tế bào gốc trung mô (MSC), hoặc sản phẩm chiết xuất từ tế bào, như túi ngoại bào từ tế bào gốc trung mô (MSC-EVs). Nó đảm bảo sự tăng trưởng và chức năng tối ưu của tế bào thông qua việc cung cấp các chất dinh dưỡng thiết yếu, yếu tố tăng trưởng và các thành phần quan trọng khác.
Việc sử dụng môi trường đạt chuẩn GMP cho nuôi cấy tế bào sẽ có những ưu điểm sau:
- Kiểm soát rủi ro từ đầu vào: Mọi thành phần trong môi trường nuôi cấy đạt chuẩn GMP phải có nguồn gốc rõ ràng và đạt chứng nhận tương ứng.
- Đảm bảo tính nhất quán giữa các lô: Việc kiểm soát chặt chẽ quy trình sản xuất GMP cho phép kiểm soát chất lượng sản phẩm môi trường nuôi cấy để đảm bảo mọi lô sản phẩm đều có chất lượng như nhau.
- Có hồ sơ tài liệu và có thể truy xuất nguồn gốc: Mọi bước trong quy trình sản xuất đều được văn bản hóa và có khả năng truy xuất nguồn gốc hoàn chỉnh.
Những ưu điểm trên của môi trường GMP giúp doanh nghiệp chuẩn hóa quy trình sản xuất và dễ dàng đáp ứng các yêu cầu khắt khe từ cơ quan quản lý trong nước lẫn quốc tế, tạo ra một bộ hồ sơ sản phẩm hoàn chỉnh, sẵn sàng cho các giai đoạn thử nghiệm lâm sàng và thương mại hóa
Kết luận
Việc chuyển dịch sang các dòng môi trường có thành phần hóa học xác định (CD) hoặc không có nguồn gốc động vật (AOF) không nên được xem là một giải pháp “vạn năng” có thể ngay lập tức giải quyết mọi thách thức trong nuôi cấy tế bào gốc trung mô (MSC) và sản xuất EV từ MSC. Tuy nhiên, đây là yêu cầu nền tảng đối với bất kỳ quy trình nào hướng tới mục tiêu sản xuất MSC-EVs đạt chuẩn sử dụng trong lâm sàng.
Việc lựa chọn môi trường nuôi cấy cần được đặt trong tư duy sản xuất, với trọng tâm là tính ổn định trong vận hành quy trình, khả năng kiểm soát rủi ro từ các yếu tố ngoại lai và độ đồng nhất của sản phẩm đầu ra. Trong lộ trình chuẩn hóa và thương mại hóa, một hệ thống nuôi cấy có thành phần xác định rõ ràng, không có nguồn gốc động vật, theo tiêu chuẩn GMP chính là nền tảng vững chắc cho sự phát triển bền vững của dự án.
Dòng môi trường CellCor™ MSC CD AOF và CellCorTM EXO CD đã được thiết kế chuyên biệt để đảm bảo tính đồng nhất tuyệt đối cho quy trình sản xuất, loại bỏ hoàn toàn các rủi ro biến thiên lô và giảm tối thiểu rủi ro nhiễm tạp.
Tìm hiểu thêm về CellCor™ MSC CD AOF
Tìm hiểu thêm về CellCorTM EXO CD
📩 Thông tin liên hệ:
- Website: https://biogroupvietnam.com/vi/lien-he/
- Hotline: +84 963 621 421
- Email: info@biogroupvietnam.com
TÀI LIỆU THAM KHẢO
- Xeno-free, Serum-free, What’s for me? – Mesenchymal Stem Cell Media [Internet]. [cited 2026 Feb 2]. Available from: https://www.rndsystems.com/blog/xeno-free-serum-free-whats-me-mesenchymal-stem-cell-media
- Culture Media Defined: Xeno-Free, Animal Origin-Free, and More | STEMCELL Technologies [Internet]. [cited 2026 Feb 2]. Available from: https://www.stemcell.com/how-do-we-define-our-media.html
- Kim Schrag, Manisha Sahni, Scott Wilson, Chas Hernandez, Andrew Christie. Strategies for Investigation of Cell Culture Media Failures: Case Study of an Insect Cell Culture Medium Failure Due to Yeastolate [Internet]. Sigma-Aldrich; [cited 2026 Feb 3]. Available from: https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/marketing/global/documents/237/010/cell-culture-media-failures.pdf
- Zheng X, Baker H, Hancock WS, Fawaz F, McCaman M, Pungor Jr. E. Proteomic Analysis for the Assessment of Different Lots of Fetal Bovine Serum as a Raw Material for Cell Culture. Part IV. Application of Proteomics to the Manufacture of Biological Drugs. Biotechnol Prog. 2006;22(5):1294–300.
- ICH Q5A(R2) Guideline on viral safety evaluation of biotechnology products derived from cell lines of human or animal origin – Scientific guideline | European Medicines Agency (EMA) [Internet]. 1997 [cited 2026 Feb 2]. Available from: https://www.ema.europa.eu/en/ich-q5ar2-guideline-viral-safety-evaluation-biotechnology-products-derived-cell-lines-human-or-animal-origin-scientific-guideline
- ICH Q7 Good manufacturing practice for active pharmaceutical ingredients – Scientific guideline | European Medicines Agency (EMA) [Internet]. 2000 [cited 2026 Feb 3]. Available from: https://www.ema.europa.eu/en/ich-q7-good-manufacturing-practice-active-pharmaceutical-ingredients-scientific-guideline
- Medicine C for V. CVM GFI #253 Current Good Manufacturing Practice for Animal Cells, Tissues, and Cell- and Tissue-Based Products [Internet]. FDA; 2022 [cited 2026 Feb 3]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cvm-gfi-253-current-good-manufacturing-practice-animal-cells-tissues-and-cell-and-tissue-based
- New guidelines on good manufacturing practices for advanced therapies | European Medicines Agency (EMA) [Internet]. 2017 [cited 2026 Feb 3]. Available from: https://www.ema.europa.eu/en/news/new-guidelines-good-manufacturing-practices-advanced-therapies
- Research C for DE and. Current Good Manufacturing Practice for Phase 1 Investigational Drugs [Internet]. FDA; 2023 [cited 2026 Feb 3]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/current-good-manufacturing-practice-phase-1-investigational-drugs
- Research C for DE and. Facts About the Current Good Manufacturing Practice (CGMP). FDA [Internet]. 2025 Nov 21 [cited 2026 Feb 3]; Available from: https://www.fda.gov/drugs/pharmaceutical-quality-resources/facts-about-current-good-manufacturing-practice-cgmp